Introduction
Cancer remains one of the greatest medical challenges of our era. While advances in genomics, immunotherapy, and targeted agents have transformed clinical outcomes in select cancers, metastasis and therapeutic resistance continue to drive mortality. The underlying biology of metastasis has been studied for decades, but the last two decades have crystallized one crucial concept: epithelial–mesenchymal transition (EMT).
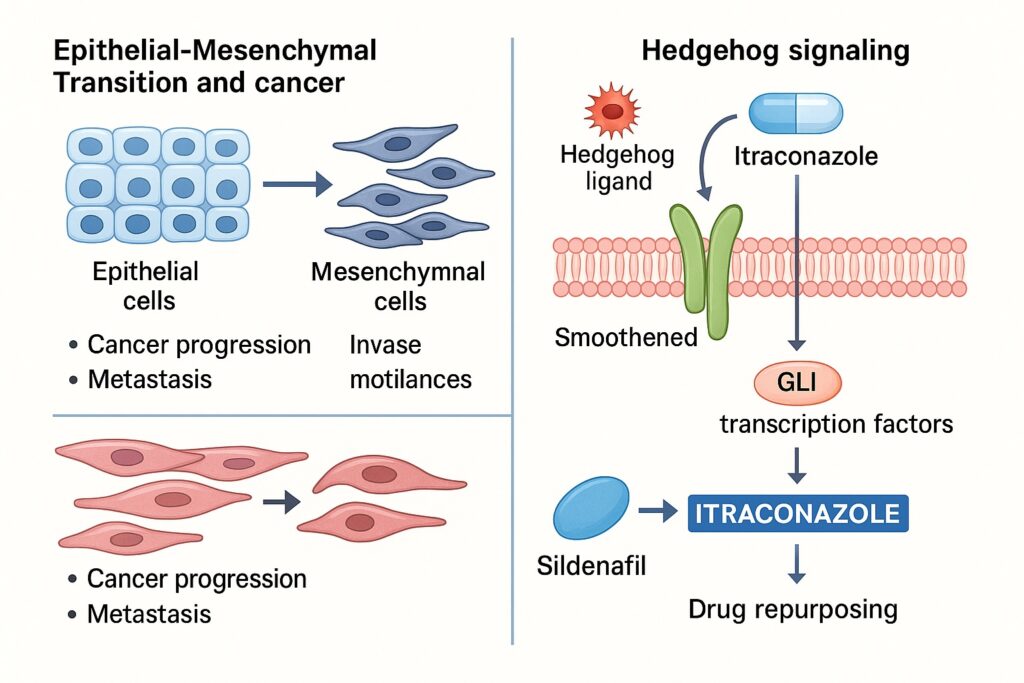
EMT is a cellular reprogramming process in which epithelial cells, typically adherent and polarized, acquire mesenchymal characteristics—motility, invasiveness, and resistance to apoptosis. Originally recognized in embryogenesis and wound healing, EMT has now been firmly implicated in cancer progression. It does not merely facilitate local invasion but also contributes to stemness, immune evasion, and drug resistance.
Targeting EMT, however, is easier said than done. This is not a single pathway but a complex reorganization of signaling cascades, transcription factors, and microenvironmental interactions. Among these, Hedgehog (Hh) signaling has emerged as a key regulator. Aberrant Hedgehog activation sustains EMT, enhances tumor-initiating capacity, and drives metastatic dissemination.
Here lies the therapeutic intrigue: a widely used antifungal drug, itraconazole, appears to inhibit Hedgehog signaling and, by extension, EMT. Repurposing itraconazole for oncology represents both an economical and mechanistically sound strategy. Remarkably, this strategy echoes another classic pharmacological tale: sildenafil, originally developed for angina, repurposed successfully for erectile dysfunction and pulmonary hypertension. In the same way, itraconazole may yet transcend its antifungal identity to become an anticancer agent.
This article examines EMT as a cornerstone of cancer progression, the role of Hedgehog signaling, and the repurposing of itraconazole as a potential therapeutic. It will also reflect on the broader principle of drug repurposing, using sildenafil as a successful precedent, and highlight the challenges and opportunities that lie ahead.
Epithelial–Mesenchymal Transition in Cancer
Defining EMT
At its core, EMT is a phenotypic plasticity program. Cells undergoing EMT lose epithelial markers such as E-cadherin, tight junction proteins, and polarity complexes. Simultaneously, they acquire mesenchymal markers including N-cadherin, vimentin, and fibronectin. This shift transforms cells from being tethered within epithelial sheets to becoming invasive migratory units.
This transformation is orchestrated by transcription factors—Snail, Slug, Twist, and Zeb1/2—that repress epithelial gene expression and activate mesenchymal genes. The process is reversible; mesenchymal–epithelial transition (MET) enables colonization at distant metastatic sites. Thus, EMT and MET represent a dynamic equilibrium central to tumor dissemination.
EMT in Metastasis
Cancer mortality is largely due to metastasis, not primary tumors. EMT facilitates local invasion by enabling tumor cells to breach basement membranes and migrate into surrounding stroma. Once intravasated into circulation, mesenchymal-like cells exhibit survival advantages, resisting anoikis and immune clearance.
At metastatic sites, MET restores epithelial traits, allowing cells to proliferate efficiently. This plasticity—switching between epithelial and mesenchymal states—makes EMT more than a linear program; it is a continuum of hybrid phenotypes, each with distinct metastatic potential.
EMT in Drug Resistance and Stemness
EMT is also implicated in therapeutic resistance. Mesenchymal-like cancer cells often upregulate drug efflux transporters, detoxification enzymes, and anti-apoptotic proteins. This makes them less susceptible to chemotherapy, radiotherapy, and even targeted agents.
Moreover, EMT endows cells with stem-like properties, increasing tumor-initiating capacity and promoting relapse after therapy. The convergence of EMT, stemness, and drug resistance explains why conventional therapies often fail to eradicate tumors completely, leaving behind resilient cell populations primed for recurrence.
Hedgehog Signaling and EMT Regulation
Overview of Hedgehog Pathway
The Hedgehog (Hh) pathway, essential in embryonic patterning, is typically quiescent in adult tissues. Its canonical cascade begins when Hh ligands (Sonic, Indian, Desert) bind to the Patched (PTCH) receptor, relieving suppression of Smoothened (SMO). Activated SMO triggers downstream activation of Gli transcription factors, which regulate genes driving proliferation, survival, and EMT.
Aberrant Hedgehog activation has been documented in basal cell carcinoma, medulloblastoma, pancreatic cancer, and more. Importantly, Hedgehog signaling intersects with EMT transcription factors, reinforcing mesenchymal traits and invasiveness.
Hedgehog and Cancer Plasticity
In tumors, Hedgehog signaling sustains cancer stem cell niches, enhances invasiveness, and promotes immune evasion. It interacts with other EMT-associated pathways—TGF-β, Wnt, Notch—creating a resilient signaling network. Hedgehog inhibition therefore represents an attractive strategy to blunt EMT and cancer progression.
Hedgehog Inhibitors in Oncology
Pharmaceutical efforts have yielded Hedgehog inhibitors such as vismodegib and sonidegib, approved for basal cell carcinoma. However, their utility beyond niche indications is limited, often due to resistance and toxicity. This has prompted interest in alternative modulators—including repurposed drugs like itraconazole.
Itraconazole: From Antifungal to Hedgehog Inhibitor
Pharmacological Background
Itraconazole, a triazole antifungal, exerts its primary action by inhibiting lanosterol 14α-demethylase, blocking ergosterol synthesis in fungal membranes. Clinically, it is widely used against systemic mycoses, with an established safety record spanning decades.
Unexpectedly, itraconazole was shown to inhibit Hedgehog signaling at the level of Smoothened. This effect is independent of its antifungal activity. Structural analyses suggest itraconazole binds distinct sites on Smoothened, altering its conformation and preventing downstream Gli activation.
Preclinical Evidence
Preclinical studies reveal itraconazole’s ability to suppress tumor growth, reduce EMT markers, and inhibit angiogenesis. In pancreatic cancer models, itraconazole reduced tumor vascularization and synergized with chemotherapy. In prostate and lung cancers, itraconazole suppressed Hedgehog signaling and decreased metastatic spread.
Importantly, itraconazole appears to reverse mesenchymal features, restoring epithelial markers and reducing invasiveness—consistent with its role as an EMT modulator.
Clinical Investigations
Clinical trials have tested itraconazole in basal cell carcinoma, prostate cancer, and non-small cell lung cancer. Results indicate measurable antitumor activity, though not yet definitive enough for regulatory approval. Ongoing studies seek to refine its dosing, optimize patient selection, and explore combinations with standard therapies.
The appeal of itraconazole lies in its repurposing potential. With known pharmacokinetics, safety, and cost-effectiveness, itraconazole bypasses many hurdles of de novo drug discovery.
Drug Repurposing in Oncology: Lessons from Sildenafil
The story of itraconazole resonates with one of the most famous examples of repurposing: sildenafil. Originally designed as an anti-anginal agent, sildenafil failed in that indication but demonstrated striking effects on penile vasodilation. Its approval for erectile dysfunction revolutionized sexual medicine, and subsequent approval for pulmonary arterial hypertension highlighted its broader vascular potential.
Like itraconazole, sildenafil’s repurposing reflects the principle that drugs often harbor unanticipated mechanisms of action. In sildenafil’s case, inhibition of PDE5 not only improved erectile function but also modulated pulmonary and systemic vascular tone. Similarly, itraconazole, by targeting Smoothened, reveals anticancer properties invisible from its antifungal role.
This parallel underscores the value of pharmacovigilance, mechanistic curiosity, and translational agility. Drug repurposing is not merely opportunistic but scientifically strategic—an efficient way to harness existing molecules for new therapeutic domains.
Challenges and Opportunities Ahead
Biological Complexity
EMT is not governed by a single pathway but by overlapping networks. Even if itraconazole suppresses Hedgehog signaling, other EMT drivers (TGF-β, Wnt, hypoxia) may compensate. Thus, monotherapy may be insufficient, necessitating rational combinations.
Clinical Translation
Itraconazole’s efficacy appears context-dependent, varying by cancer type and genetic background. Biomarkers predicting Hedgehog dependence are essential for selecting responsive patients.
Safety and Dosing
Although itraconazole is generally safe, chronic high-dose use may pose hepatotoxicity and drug–drug interactions. Oncology trials must navigate these issues carefully, especially in patients on polypharmacy.
Future Horizons
The integration of artificial intelligence and systems biology may accelerate identification of repurposing candidates, optimize combinations, and predict toxicity. Itraconazole could serve as a prototype for future repurposed EMT inhibitors, guiding a new wave of economical oncology innovation.
Conclusion
Epithelial–mesenchymal transition is the biological linchpin of cancer metastasis, stemness, and therapeutic resistance. Hedgehog signaling, as one of its key regulators, represents a tractable target. The antifungal itraconazole, through Hedgehog inhibition, offers a promising avenue to blunt EMT and restrain cancer progression.
The broader lesson is clear: drug repurposing is not a side story but a powerful paradigm in oncology. Just as sildenafil found new life beyond its intended purpose, itraconazole may yet be enlisted against one of medicine’s greatest foes. Success will require rigorous trials, biomarker-driven patient selection, and perhaps, a little pharmacological serendipity.
FAQ
1. What is EMT and why is it important in cancer?
EMT is a process where epithelial cells acquire mesenchymal traits, becoming more invasive and resistant to therapy. It is central to metastasis and recurrence.
2. How does itraconazole affect cancer cells?
Itraconazole inhibits Hedgehog signaling, reducing EMT markers, angiogenesis, and tumor growth in preclinical and early clinical studies.
3. Why mention sildenafil in this context?
Sildenafil exemplifies successful drug repurposing. Its story parallels itraconazole’s potential journey from antifungal to anticancer therapy.
4. What are the main challenges of using itraconazole in oncology?
Challenges include biological redundancy of EMT pathways, identifying responsive patients, managing toxicity, and proving efficacy in large clinical trials.
References
- Nieto MA, Huang RYJ, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45.
- Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18(2):128–134.
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15(23):3059–3087.
- Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with Hedgehog pathway inhibitor vismodegib. N Engl J Med. 2009;361:1173–1178.
- Kim J, Tang JY, Gong R, et al. Itraconazole, a commonly used antifungal, targets Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17(4):388–399.
- Antonarakis ES, Heath EI, Smith DC, et al. Repurposing itraconazole as a treatment for prostate cancer. Oncologist. 2013;18(2):163–173.
- Pantziarka P, Bouche G, Meheus L, Sukhatme V, Sukhatme VP. Repurposing drugs in oncology (ReDO)—itraconazole as an anti-cancer agent. Ecancermedicalscience. 2015;9:521.
- Corbin JD, Francis SH. Pharmacology of phosphodiesterase-5 inhibitors. Int J Clin Pract. 2002;56(6):453–459.
- LoMonte G, Pizzuti A, et al. Drug repurposing in oncology: opportunities and challenges. Cancers (Basel). 2020;12(10):2909.